
Hjælpemidler
Hjælpemidler skal opfylde krav til medicinsk udstyr.
Hjælpemidler er med til at behandle, forebygge og lindre sygdom hos mennesker og betragtes derfor som medicinsk udstyr.
En hjælpemiddel producent kan CE mærke sit produkt, hvis det kan dokumenteres at produktet lever op til kravene i Direktivet for medicinsk udstyr.
Direktivet for medicinsk udstyr er implementeret i dansk lov gennem bekendtgørelse 1263 af 15. december 2008 om medicinsk udstyr. Bekendtgørelsen ligger under Ministeriet for Sundhed og Forebyggelse, og efterlevelse af reglerne varetages af Sundhedsstyrelsen i den afdeling som hedder Medicinsk udstyr.
Når et hjælpemiddel skal opfylde krav til CE mærkning, er det således efter retningslinier i bekendtgørelse 1263:2008.
Krav som skal opfyldes fremgår i vidt omfang af bekendtgørelsens bilag I Væsentlige krav, hvor der listes en lang række forhold som skal være opfyldt.
I forhold til at opfylde mange af disse krav, henviser bekendtgørelsen til, at man skal efterleve relevante gældende harmoniserede standarder. Det betyder i praksis, at hvis der i forhold til et produkt er beskrevet en testmetode, krav til sikkerhed eller andet, så vil man skulle anvende og opfylde krav i disse standarder. Hvis man vælger noget andet, skal man som fabrikant kunne dokumenter produktets sikkerhed og sundhed, ligesom man skal kunne forklare, hvorfor man har valgt en anden test og analyse metode/ standard. Også generelle standarder som er udarbejdet , for eksempel i forhold til grafiske symboler, risikoanalyse, biokompabilitet og lignende, vil man som udgangs punkt forvente bliver anvendt og efterlevet, inden man som fabrikant, sætter CE mærke på sit produkt.
DS/EN 12182:2012
l forhold til hjælpemidler er en af de standarder man skal have fat i, DS/EN 12182:2012 “Hjælpemidler til personer med funktionsnedsættelse – Generelle krav og prøvningsmethoder”
Standarden DS/EN 12182 er at regne for det som man kalder en ”niveau 1 ” standard.
Der er 3 niveauer af europæiske standarder for tekniske hjælpemidler til personer med funktionsnedsættelse. Disse er :
Niveau 1 : Gennerelle krav til tekniske hjælpemidler
Niveau 2 : Særlige krav til produktgrupper af tekniske hjælpemidler
Niveau 3 : Specifikke krav til typer af tekniske hjælpemidler
Niveau 1 er det højeste, mens Niveau 2 og 3 kan kombineres i et enkelt dokument.
Hvor der forefindes standarder for særlige hjælpemidler eller grupper af hjælpemidler (niveau 2 eller 3) kan DS/EN 12182 ikke stå alene, og fabrikant vil i forhold til at opfylde krav til CE mærkning skulle forholde sig til disse standarder på lavere niveauer.
Andre relevante standarder og guidelines.
Herunder er givet eksempler på andre standarder og guidelines som kan være relevante. Listen er ikke udtømmende og udelukkende af vejledende karakter.
DS/EN ISO 14971 Medicinsk udstyr – håndtering af risikostyring for medicinsk udstyr, beskriver krav til risikohåndtering og metoder
”MEDDEV 2.7.1 Evaluation Of Clinical Data: A Guide For Manufacturers And Notified Bodies”.
DS/ EN 60601-1 Elektromedicinsk udstyr del 1 : Generelle sikkerhedskrav og væsentlige funktionskrav.
DS/EN 62304 Software for medicinsk udstyr – Livscyklus processer for software
” General principles of software validation ; Final guidance for industry and FDA staff” (2002).
Maskindirektiv 2006/42/EF er trådte i kraft d. 29. december 2009. Direktivet er implementeret i dansk lovgivning via Arbejdstilsynets bekendtgørelse nr. 612 af 25. juni 2008 om indretning af tekniske hjælpemidler, kapitel 2.
DS/EN ISO 10993-1 : 2009 Biologisk vurdering af medicinsk udstyr – Del 1: Vurdering og prøvning indenfor rammerne af et risikoledelsessystem.
Opfyldelse af krav :
Ved opfyldelse af krav i lovgivning og standard, er følgende krav generelle og bærende for alt medicinsk udstyr/ hjælpemidler :
1.Risikoanalyse
2.Klinisk evaluering
Risikoanalyse.
Risikoanalyse skal vurdere fare og risici i alle processer og i hele produktets levetid. Ved risikoanalyse, er det således nødvendigt at kigge bredt lige fra design og til bortskaffelse. Eksempler på processer som bør være beskrevet i risikoanalyse : design, tilsigtet ydeevne, målgruppe, materiale, indkøb, fremstilling, frigivelse/ godkendelse, mærkning, pakning og forsendelse, modtagelse hos kunde, dokumentation, anvendelse og bortskaffelse. (listen er kun tænkt som eksempel, den kan se ud på mange andre måder, og med andre punkter).
Systematisk metode.
Standarden DS/EN ISO 14971 ”Medicinsk udstyr – håndtering af risikostyring for medicinsk udstyr”, beskriver krav til risikohåndtering og metoder.
Figur 1 i DS/EN ISO 14971 beskriver retningslinier i forhold til risikohåndterings processen.
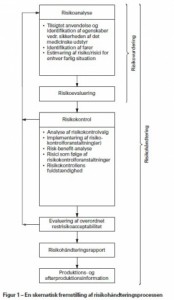
Med udgangs punkt i figur 1 herover, vil man som fabrikant skulle udarbejde en kort beskrivelse af, hvordan man som virksomhed sikre at risikoanalyse gennemføres og håndteres. Der er forskellige metoder som kan anvendes til selve analysen, – FMEA (failure modes and effects analysis) er en af de metoder som ISO 14971 anbefaler.
Ved analyse af risici, tages der udgangspunkt i det enkelte produkt (eller hvis flere produkter er ”ens” kan man lave risikoanalyse for en produktgruppe), og ud fra det, udarbejde en oversigt over de risici som kan optræde, eller som man via særlig procedure og kontroller har forhindret i at optræde.
Risikoanalysen er et ”levende” dokument som fabrikant skal opdatere og følge løbende, eksempelvis ved ændring af produkt, henvendelser fra brugere vedrørende fejl eller risici på produkt, som man ikke allerede har håndteret i risikoanalysen. Risikoanalysen er, hvis der opstå problemer/ hændelser, et af fabrikantens vigtigste dokumenter, i forhold til at bevise, at han/ hun har efterlevet gældende lov, og ikke sendt sundheds og sikkerhedsmæssigt farlige produkter på markedet.
Vudering af den enkelte risici.
I ISO 14971 er beskrevet en metode til hvordan man evaluere hvert enkelt punkt i risikoanalysen.
Se figur B1.
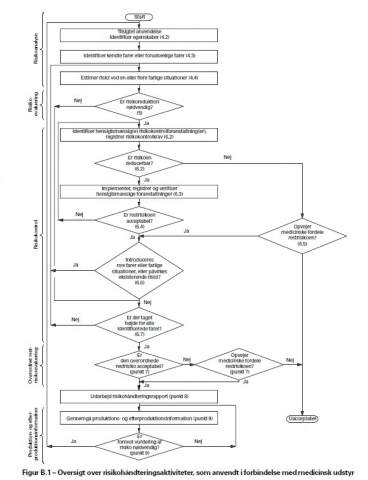
Som det fremgår af figur B1, er der en kasse som hedder ”opvejer medicinsk fordele restrisiko ? ”.
Svare man ja, fortsætter man videre frem mod udarbejdelse af endelig rapport, mens man hvis man svare nej, og ender i ”Uacceptabel” , må til at finde ud af hvad man kan gøre for at minimere risiko.
Det interessante er, at man godt kan sende et ”farligt” produkt på markedet, – hvis den sygdom/ lidelse udstyret er tiltænkt anvendt til, har en sådanne karakter at man hellere vil løbe en kendt risiko, end helt at undvære udstyret. Forholdsregler, kendte risici og sundhedsmæssige bivirkninger vil dog altid skulle kommunikeres til bruger/ behandler.
Klinisk evaluering .
Klinisk evaluering er et krav i såvel DS/EN 12182:2012 som i bekendtgørelsen om medicinsk udstyr 1263 af 15. dec. 2008.
I bekendtgørelsens Bilag I Væsentlige krav punkt 6 a står der :
” Påvisningen af overensstemmelse med de væsentlige krav skal omfatte en klinisk evaluering i overensstemmelse med bilag X.”
En klinisk evaluering kan enten foretaget ved :
a.evaluering af relevant, foreliggende videnskabelig litteratur, som beskriver udstyrets sikkerhed, ydeevne konstruktionskarakteristika og formål, (den af fabrikant anførte ydeevne) , samt en vurdering af om bivirkninger er acceptable. Anvendt litteratur skal være passende/ relevant i forhold til at kunne sammenligne med det medicinsk udstyr som ønskes evalueret.
b.klinisk afprøvning, med det formå :
– at kontrollere at udstyrets ydeevne under normal anvendelsesforhold svare til den
– at fastslå eventuelle bivirkninger og uønskede følgevirkninger under normal anvendelse,
og vurdere om de udgør en risiko sammenlignet med udstyrets angivne ydeevne.
Klinisk afprøvning skal anmeldes til Lægemiddelstyrelsen, og man skal have tilladelse inden man går i gang.
Klinisk evaluering via litteraturstudier, skal følge retningslinier i EU-Kommissionens vejledning om evaluering af kliniske data ”MEDDEV 2.7.1 Evaluation Of Clinical Data: A Guide For Manufacturers And Notified Bodies”.
Følgende systematik for klinisk evaluering via litteratur studier er beskrevet i MEDDEV 2.7.1 rev. 3
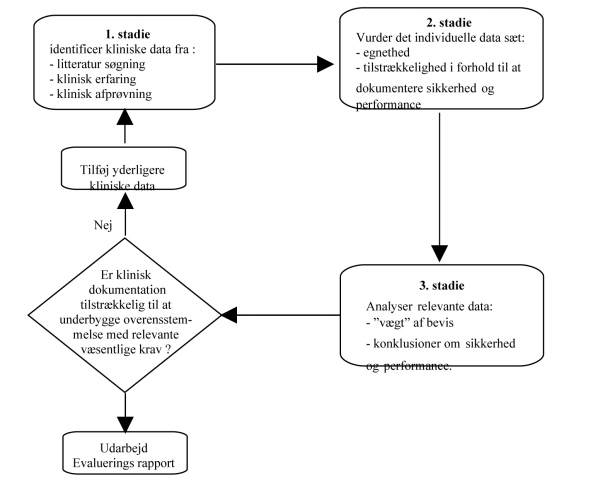
Litteratur søgning er dog ikke så ligetil, – der er krav om at man på ”videnskabelig vis” skal kunne dokumenter søgning, hvad der søges ud fra, hvilken litteratur der vælges til og vælges fra, og at man evaluere den litteratur som er fundet egnet. Alt dette gør man jo i takt med at man finder litteraturen og læser den, men hvad langt de fleste ikke får gjort, er at dokumentere ( skrive ned) at det er gjort, og hvordan det er gjort.
Det er således ikke nok, at man kan vise hvilken dokumentation man har valgt til, man skal også kunne forklare hvad man har valgt fra !
MEDDEV 2.7.1 rev 3. beskriver følgende retningslinier for litteratur søgning.
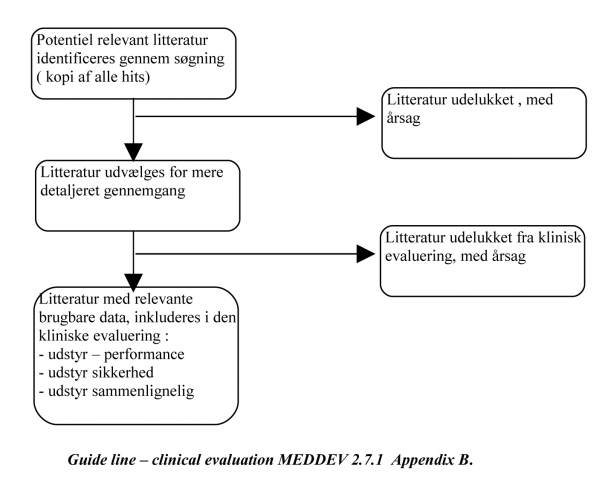
Guide line – clinical evaluation MEDDEV 2.7.1 Appendix B.
Befæstigelse, rengøring, støj m.m.
Standarden DS/EN 12182:2012 fortsætter med krav i forhold til om hjælpemidler kan skilles ad, engangsbefæstelseselementer, brugervægt, antændelighed af materialer, madrasser og sengeudstyr, biokompatibilitet og toksicitet, rengøring , korrosionsbestandighed, støj og vibrationer.
Elektronisk udstyr – et afsnit for sig.
Mere og mere medicinsk udstyr/ hjælpemidler indeholder elektronik. Produkter som indeholder elektriske eller elektronisk udstyr / komponenter skal være opmærksomme på yderligere krav i forhold til dette. Dels de ”almindelige ” lovkrav vedrørende elektrisk udstyr som eksempelvis lavspændings direktivet , men også den særlige standard for medicinsk udstyr 60601 serien, hvor der stilles krav til EMC , el-sikkerhed, faldtest, mærkning, alarm signaler og lignende.
Er der i udstyr givet mulighed for anvendelse af batterier, skal man være opmærksom på at også denne facilitet skal være baseret på risikoanalyse, som identificere fare og eventuelle risici forbundet hermed. (udslip af syre og andre stoffer fra batteri, kortslutning og lignende).
Software.
Software og medicinsk udstyr er igen et kapitel for sig. Kravene siger at software skal valideres. Det vil sige at man gennem en dokumenteret metode, sikre sig at programmer og eventuelle beskeder på skærm, altid gør som tiltænkt. Standarden DS/EN 62304:2006 ”Software for medicinsk udstyr – livscyklusprocesser for software” beskriver hvordan man kan opbygge et system, som tager hånd om software i hele udstyrets levetid.
FDA – den Amerikanske myndighed som er ansvarlig for godkendelse af medicinsk udstyr/ hjælpemidler til det Amerikanske marked, har udgivet en guideline som man, hvis man skal sælge til USA, skal være opmærksom på.
” General principles of software validation ; Final guidance for industry and FDA staff” (2002).
Nogle af de ting man som fabrikant skal være opmærksom på ved software validering, er at udarbejde en plan for hvordan man godkender software, – hvilke tests der skal gennemføres, hvornår, hvem er ansvarlig, documentation for resultat af tests, hvad gør man hvis der er fejl, evaluering og godkendelse af software samt versions styring. En samlet beskrivelse af disse forhold, skabeloner til tests og/ eller leverandør aftaler om tests vil være et minimum, i forhold til produkter med software.
Ved test af software, skal der tænkes i hele produktets levetid, versions styring, opdatering og ændring af versioner, stress test, provokerede fejlmuligheder, brugervenlighed, afprøvning i tiltænkt miljø og med tiltænkt målgruppe for at nævnt nogle eksempler.
Bevægelige dele.
I forhold til hjælpemidler er man underlagt almindelige regler for maskinsikkerhed, og skal sikre at der ikke er risiko for eksempelvis klemning.
Maskindirektiv 2006/42/EF er trådte i kraft d. 29. december 2009. Direktivet er implementeret i dansk lovgivning via Arbejdstilsynets bekendtgørelse nr. 612 af 25. juni 2008 om indretning af tekniske hjælpemidler, kapitel 2.
Hjælpemidler som er en ”maskine” (groft oversat til ”bevægelige dele” ), er underlagt såvel direktiv om medicinsk udstyr, som maskindirektivet.
Direktivet om medicinsk udstyr er ”hoveddirektiv” og retningsgivende, men for at opfylde krav i direktivet´s bilag I Væsentlige krav, bliver man nød til at ”dække sig ind” , ved at opfylde andre direktiver hvor det er relevant, det kan være maskindirektivet, eller andre produktspecifikke standarder som er udviklet.
Eksempelvis er der i forhold til kørestole udviklet en lang række specifikke standarder. (ISO 7176 serien med flere), ligesom også hospitalssenge har ”deres egen” standard,- IEC 60601-2-52 ”særlige krav til grundlæggende sikkerhed for hospitals- og plejesenge”.
Oversigt over standarder med relation til kørestole kan findes her:
http://www.iso.org/iso/iso_catalogue/catalogue_tc/catalogue_tc_browse.htm?commid=53792&published=on
På EU´s hjemmeside for maskindirektivet kan man finde en oversigt over standarder med relation til maskindirektivet.
http://ec.europa.eu/enterprise/sectors/mechanical/documents/standardization/machinery/harmonised/
Fabrikantens oplysninger.
Fabrikantens oplysninger kan opdeles i to hovedemner :
1.Mærkning
2.Brugsanvisning
Mærkning:
Krav til mærkning er beskrevet i bekendtgørelse om medicinsk udstyr Bilag I, punkt 13.3
13.3. Mærkningen skal omfatte følgende oplysninger:
a) fabrikantens navn eller firmanavn og adresse. For udstyr, som importeres til Fællesskabet med henblik på distribution i Fællesskabet, skal mærkningen eller den ydre emballage eller brugsanvisningen desuden angive repræsentantens navn og adresse, hvis fabrikanten ikke har noget hovedsæde i Fællesskabet
b) de angivelser, som er absolut nødvendige, for at brugeren kan identificere udstyret og emballagens indhold
c) betegnelsen “STERILE”, hvis dette er relevant
d) i givet fald betegnelsen “LOT” efterfulgt af batchkoden eller serienummeret
e) i givet fald den dato, frem til hvilken det er fuldt forsvarligt at anvende udstyret, angivet med år og måned
f) i givet fald angivelse af, at udstyret er beregnet til engangsbrug. En fabrikants angivelse af engangsbrug skal være konsekvent i hele Fællesskabet
g) for udstyr efter mål påtegningen “specialfremstillet udstyr”
h) for udstyr bestemt til klinisk afprøvning, påtegningen “udelukkende til klinisk afprøvning”
i) særlige betingelser vedrørende opbevaring og/eller håndtering
j) særlige brugsanvisninger
k) advarsler og/eller forholdsregler
l) for andet aktivt udstyr end det, der hører under litra e), fremstillingsår. Denne oplysning kan indgå i batch- eller serienummeret
m) steriliseringsmetode, hvis en sådan er relevant.
n) For udstyr som omhandlet i § 1, stk. 5, skal mærkningen angive, at udstyret som en integreret bestanddel indeholder et stof fremstillet af humant blod.
13.4. Såfremt et udstyrs formål ikke er indlysende for brugeren, skal fabrikanten klart angive formålet på mærkningen og i brugsanvisningen.
13.5. Udstyr og aftagelige bestanddele skal, hvis dette er rimeligt og muligt, identificeres, i givet fald i form af batchnummer, således at det bliver muligt at træffe de relevante foranstaltninger til at erkende en potentiel risiko i forbindelse med udstyret og de aftagelige bestanddele.
Oplysninger skal anføres i form af symboler og vejledende forklaring.
Alle symboler eller identifikationsfarver skal være i overensstemmelse med harmoniserede standarder. Hvis der ikke findes nogen standard på det pågældende område, skal symboler og farver være beskrevet i den dokumentation, som ledsager udstyret.
ISO 980:2008 ”Grafiske symboler til anvendelse ved medicinsk udstyr”, er en af de standarder som gælder for praktisk taget alt medicinsk udstyr/ hjælpemidler. Derudover kommer produkt specifikke krav til symboler, for eksempel for elektrisk udstyr er der en række yderligere symboler i forhold til el- sikkerhed som skal tages med.
Brugsanvisning.
Krav til brugsanvisning fremgår af bekendtgørelsen om medicinsk udstyr bilag I , punkt 13.1 og 13.6.
13.1.
Alt udstyr skal emballeres sammen med en brugsanvisning. En sådan er undtagelsesvis ikke nødvendig for udstyr i klasse I og klasse IIa, hvis det kan anvendes fuldstændig sikkert uden hjælp af sådanne anvisninger.
13.6. Brugsanvisningen skal i givet fald omfatte følgende:
a) de angivelser, der er omhandlet i punkt 13.3, med undtagelse af litra d) og e)
b) oplysning om den ydeevne, der er omhandlet i punkt 3, samt om eventuelle uønskede bivirkninger
c) for udstyr, der skal installeres sammen med eller tilsluttes andet medicinsk udstyr for at kunne fungere i overensstemmelse med dets formål, sådanne oplysninger om dets karakteristika, som er nødvendige for at kunne identificere det korrekte udstyr, som skal anvendes for at opnå en sikker kombination
d) alle oplysninger, der er nødvendige for at kontrollere, om et udstyr er installeret korrekt og kan fungere korrekt og sikkert, samt oplysninger om, hvilke vedligeholdelses- og kalibreringsforanstaltninger der skal gennemføres, og hvor hyppigt dette skal ske for at sikre, at udstyret til enhver tid fungerer korrekt og sikkert
e) i givet fald oplysninger med henblik på at undgå visse risici i forbindelse med implantation af udstyret
f) oplysninger om faren for gensidig interferens som følge af udstyret ved specifikke undersøgelser eller behandlinger
g) de nødvendige anvisninger i tilfælde af brud på den sterile emballage og i givet fald oplysninger om passende gensteriliseringsmetoder
h) for genanvendeligt udstyr oplysninger om, hvilke metoder der bør anvendes, for at genanvendelse kan finde sted, herunder rensning, desinfektion, emballering og i givet fald steriliseringsmetode, hvis udstyret skal steriliseres på ny, samt oplysning om enhver restriktion med hensyn til det mulige antal genanvendelser.
hvis der er tale om et klasse I eller IIa udstyr, og udstyrets anvendelse er åbenlys, og produkt ikke kan anvendes forkert, er det ikke et krav at der skal udarbejdes en brugsanvisning. I alle andre tilfælde vil der skulle udarbejdes en brugsanvisning som skal vedlægges varen.
Vær også opmærksom på at, hvis udstyr leveres således at det skal steriliseres før anvendelsen, skal rengørings og steriliseringsanvisning være vedlagt varen.
Hvis udstyr er beregnet til engangsbrug, skal der vedlægges oplysninger om de kendte karakteristika og tekniske faktorer, som fabrikant har kendskab til kan udgøre en risiko, hvis udstyr genanvendes.
Hvis der ikke er vurderet at være behov for brugsanvisning, skal oplysninger være tilgængelige for brugeren efter anmodning.
Brugsanvisning skal være versionsstyret, – det vil sige at der skal være en udstedelsesdato eller dato for seneste revision af brugsanvisning. (eller et revisions nummer, som fabrikant så kan fortælle hvornår er blevet godkendt).
ISO 12182:2002 har nogle ekstra ”hjælpende” krav/ punkter man skal forholde sig til for eksempel:
Nedsat syn:
”oplysninger som gælder for og udleveres sammen med hjælpemidler, som fra fabrikantens side er beregnet til anvendelse for personer med nedsat syn, skal være i følbar (fx braille) eller hørbar form” (22.2)
Støj:
” hvis et hjælpemiddel kan forårsage støjrisici, skal der gives advarsler og råd om forholdsregler vedrørende høje lydniveauer” (22.9)
Overensstemmelseserklæring.
Med opfyldelse af alle de væsentlige krav i bekendtgørelse, er man som fabrikant nu klar til at udarbejde en Overensstemmelseserklæring / Declaration of conformity .
I overensstemmelseserklæringen skriver man hvilke produkt (produktgruppe) det gælder for, hvilke standarder, lovgivning og guidelines der er efterlevet, fabrikant navn og adresse , dato og underskrift.
Overensstemmelseserklæring og alle de dokumenter der ligger som dokumentation, gemmes samlet i en eller to mapper, eller der skrives henvisning til hvor original dokumenter opbevares.
Eksportcertifikat.
Vil man eksportere til lande udenfor EU, kan man rette henvendelse til Lægemiddelstyrelsen og bede om at få et eksportcertifikat. Det er gratis, og udstedes til fabrikanter af medicinsk udstyr, som har hovedsæde i Danmark.
Man kan læse mere om dette på lægemiddelstyrelsens hjemmeside. https://laegemiddelstyrelsen.dk/da/udstyr/eksportcertifikat
Definitioner – Hjælpemidler:
DS/ EN 12182:2012
I standarden DS/ EN 12182:2012 ”Tekniske hjælpemidler til handicappede” er Tekniske hjælpemidler defineret således :
”Instrument, udstyr (til personer med funktionsnedsættelse) eller teknisk system , som af fabrikanten er beregnet til anvendelse som forebyggelse, behandling eller lindring af eller kompensation for skade, funktionshæmning, funktionsnedsættelse eller handicap”.
Bekendtgørelse 1263/2008 om medicinsk udstyr :
Bekendtgørelsen om medicinsk udstyr skriver i Kapitel 1, § 1
1) “Medicinsk udstyr”: Ethvert instrument, apparat, udstyr, software, materiale eller anden genstand anvendt alene eller i kombination, herunder software, som af fabrikanten er beregnet til specifik anvendelse til diagnostiske eller terapeutiske formål, og som hører med til korrekt brug heraf, og som af fabrikanten er beregnet til anvendelse på mennesker med henblik på:
a) Diagnosticering, forebyggelse, overvågning, behandling eller lindring af sygdomme,
b) diagnosticering, overvågning, behandling, lindring af eller kompensation for skader eller handicap,
c) undersøgelse, udskiftning eller ændring af anatomien eller en fysiologisk proces, eller
d) svangerskabsforebyggelse,
og hvis forventede hovedvirkning i eller på det menneskelige legeme ikke fremkaldes ad farmakologisk, immunologisk eller metabolisk vej, men hvis virkning kan understøttes ad denne vej.
Den Nordamerikanske definition
I Nordamerika arbejder man med en meget bred, helhedsorienteret definition:
“(…) udstyr eller systemer, hvad enten det er almindelige produkter, tilpassede eller særlige produkter, som har til formål at øge eller forbedre muligheder for aktivitet og deltagelse hos personer med funktionsnedsættelser”
“Hjælpemiddelformidling: Enhver serviceydelse, som direkte hjælper personer med nedsat funktionsevne med at udvælge, anskaffe eller anvende et hjælpemiddel.”
(ref. Hjælpemiddelinstituttet)
ISO 9999
Standard DS/ISO 9999, “Hjælpemidler til personer med funktionsnedsættelse”:
“Ethvert produkt (herunder genstande, udstyr, redskaber, teknologi og software) – specielt produceret eller almindeligt tilgængeligt, som forhindrer, kompenserer, kontrollerer letter eller neutraliserer funktionsnedsættelser, aktivitetsbegrænsninger og/eller deltagelsesbegrænsninger.”

Kurser

Kvalitetsstyring

Home

CE- mærkning af medicinsk udstyr